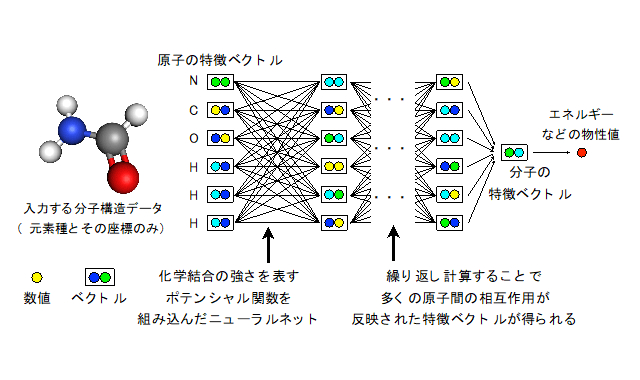
物理化学の知識を組み込んだ機械学習手法
産業技術総合研究所(産総研)は2018年9月19日、東京大学の生産技術研究所と共同で、物理化学の知識と機械学習を組み合わせて、化学物質の分子構造から物性値を高速・高精度に予測する手法を開発したと発表した。理論計算を1万倍以上高速化できると説明している。
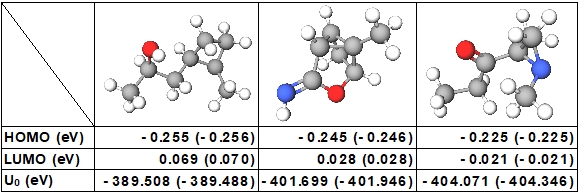
機械学習で予測された物性値。カッコ内は理論計算で得られた値
アメリカ化学会の化学物質データベースに登録されている化学物質の種類は、約3000万種に及ぶという。化学物質には光の吸収や熱伝導などの強弱などの物性値があり、構成する元素の種類や分子の構造、化学結合の強さなどによって物性値は決まってくる。
これまでは物性値を確認するため、実際に物性値を測定するか、化学物質の分子構造をコンピューターで理論計算するか、どちらかの手段が採られていた。しかし、どちらも設備や専門知識、経験が必要となり、長い時間もかかっていた。
そうした背景から、分子構造のデータをそのまま入力して物性値を予測する柔軟な機械学習の手法が研究されるようになっている。しかし、従来の手法では学習結果の解釈が難しく、学習結果の妥当性を検証できずにいた。
そこで産総研は、物理化学の知識を利用して、機械学習によって化学物質の物性値を予測する方法の研究に着手。物理化学の分野で用いられている近似式に基づいて、分子中の原子間に、化学結合などの相互作用の「強さの変化」を「バネの伸び縮み」で表すような関数を設定した。その関数を表す機械学習モデルを、大規模データで学習させる手法を編み出した。この関数は、原子間の相互作用や化学結合の強さに対応するため、学習結果の物理化学的な解釈・検証が可能だという。
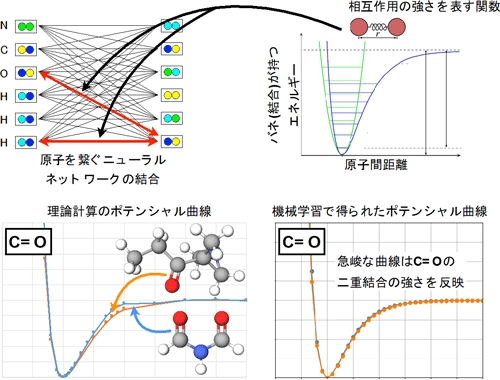
化学結合の強さを示すポテンシャル曲線(左が理論計算、右が機械学習で得られた結果)
今回考案した手法を用いて、化学物質13万種類以上が登録された大規模データベースを学習させ、予測の計算時間と精度を評価。理論計算と同じくらい高精度に、分子が示すさまざまな物性値を理論計算の1万倍以上の速さで予測できると確認した。
さらに、原子間の化学結合の強さを示すポテンシャルについて、物理化学の理論計算によって得られたものと、今回の手法によって得られたものとを比較・検証した。すると、それらが良く一致すると確認できた。今回の手法が、物理化学的に解釈できる情報をデータから学習したことを示すことになるとしている。