- 2024-8-30
- 化学・素材系, 技術ニュース, 海外ニュース
- 3次元ユークリッド群, e3nn(3D Euclidean neural network), Frontier, INCITEプログラム, MIT, Proceedings of the National Sciences, SRO(short-range order):短距離秩序, アメリカエネルギー省, アモルファス合金, ニューラルネットワーク, 世界最速スーパーコンピューター, 化学モチーフ(chemical motif), 原子規則配列, 合金成分原子, 固溶状態, 学術, 機械学習技術, 金属間化合物, 高エントロピー合金
Credits:Image courtesy of the researchers.
マサチューセッツ工科大学(MIT)は2024年7月18日、同大学の研究チームが機械学習技術を用いて、合金の固溶状態における局所的な原子規則配列「SRO(short-range order:短距離秩序)」を定量予測する手法を考案したと発表した。合金の固溶状態においても存在する、特定成分間で近接し合う傾向を表すSROについて、成分元素の電子構造計算に基づいて得られる化学結合状態と、数十億の原子規則配列である「化学モチーフ(chemical motif)」のデータベースを機械学習により関係付けて、合金のSROを特定するものだ。研究チームは、新しいカテゴリーの金属として関心を集めている多種主要元素型の高エントロピー合金の機械的および熱力学的な解析を促進するツールになると期待している。
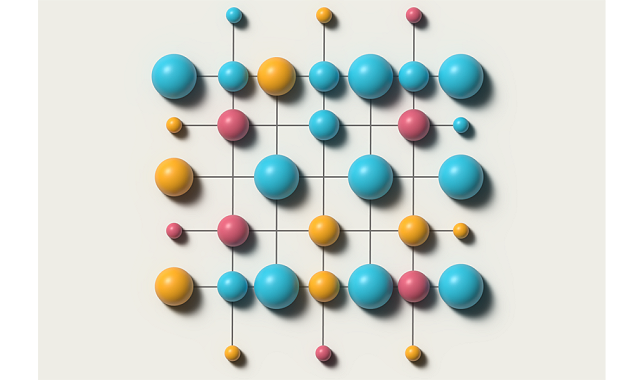
合金、特に固溶型の合金における成分元素の分布は、一般にランダムに配列しているように認識されているが、実際には「合金成分原子は、特定の他種類の成分原子と近接し合う傾向を持つ。すなわち、成分元素の分布は完全にランダムではなく、局所的な原子規則配列SROが存在する」と、研究チームは語る。しかし、SROを実験的に把握することが難しいことに加え、想定される原子規則配列である化学モチーフの種類が膨大であることから、SROを理論的に定量予測するのは簡単なことではない。そのため、これまでの材料科学において、SROが詳細に探求されることはなかった。
一方で、多種主要元素型の高エントロピー合金が、新しいカテゴリーの金属として関心を集めるようになり、高強度や高耐食性など高機能な高エントロピー合金を設計するために、SROを解読することが必要になってきている。SROを特定する伝統的な方法は、原子数を制限した小さな計算モデルに基づくシミュレーションによるものだが、少数の原子だけでは十分なシミュレーションができず、高エントロピー合金のような複雑な材料系について不完全な姿しか提供できない。研究チームは「十分な原子数を用いた高レベルの計算モデルが必要だ」と指摘する。
このような伝統的な方法の欠点を克服するために、研究チームは機械学習に着目した。最初に、成分原子の電子構造に基づいて高エントロピー合金の化学結合状態を再現するモデルを構築した。次に、データベースにある数十億個の化学モチーフについて、回転対称、鏡面対称、反転対称の関係にある等価なモチーフが現れるためシミュレーションで予測するのが困難だという課題を、こうした対称性を備えた3次元ユークリッド群を扱うニューラルネットワークである「e3nn(3D Euclidean neural networks)」を利用することで特定に成功した。最終的に、SROが未知の合金について、化学結合状態と化学モチーフのデータベースを機械学習により関係付けて、SROを特定し定量予測するフレームワークを実現した。
こうした一連の取り組みにより、局所的な結晶格子の歪みなど実験測定値と整合するSROを予測できるようになった。研究チームは、アメリカエネルギー省のINCITEプログラムにある世界最速スーパーコンピューター「Frontier」を用いて、開発したフレームワークを検証する計画だ。従来型合金、金属間化合物、アモルファス合金などとは異なる、新しい高エントロピー合金の研究を促進することが期待される。
研究成果は、2024年6月13日に『Proceedings of the National Sciences』誌に公開されている。